再生医療等製品の製造現場において、交叉汚染の防止と品質の恒常性を担保するために、洗浄バリデーションは極めて重要なプロセスです。特に新規ラインの立ち上げや査察対応を控えている製造管理者や品質保証担当者の方々にとって、GCTP省令や関連ガイドラインに準拠した「洗浄バリデーションの実施要領」を確立することは、喫緊の課題ではないでしょうか。
本記事では、再生医療分野特有の要件を踏まえ、科学的根拠に基づいた残留許容値の設定から、具体的な実施手順、サンプリング手法の選定までを網羅的に解説します。規制要件を満たし、実務で確実に運用できる洗浄バリデーション計画の策定にお役立てください。
再生医療における洗浄バリデーション実施要領の全体像
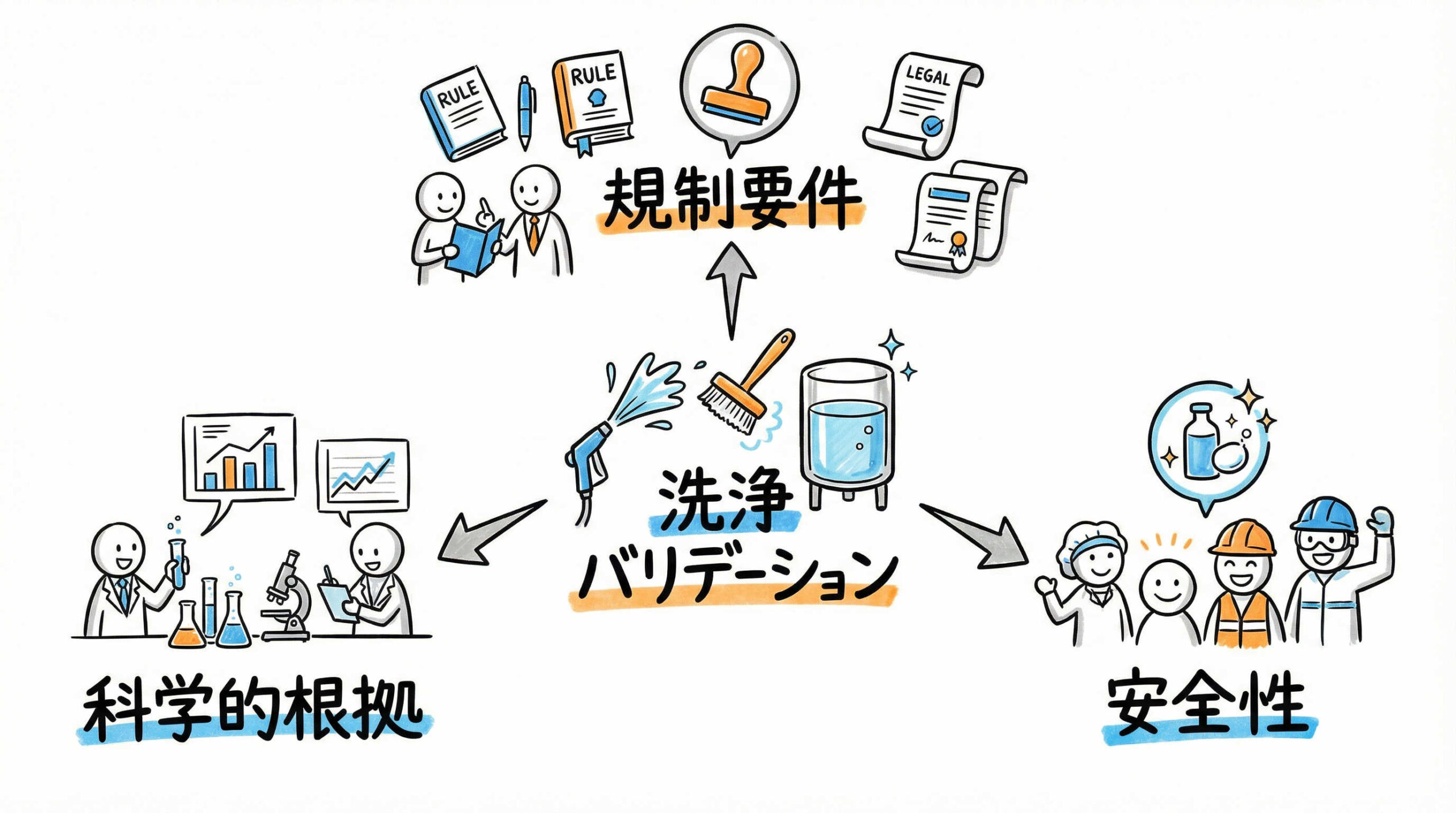
再生医療等製品の製造において、洗浄バリデーションは単なる清掃確認ではなく、製品の安全性と有効性を保証するための科学的な検証プロセスです。ここでは、GCTP省令等の規制要件が求める本質的な目的と、医薬品GMPとの相違点、そして汚染管理戦略全体における位置づけについて整理します。全体像を把握することで、より的確な実施要領の策定が可能になります。
GCTP省令が求める洗浄バリデーションの目的と定義
GCTP省令(再生医療等製品の製造管理及び品質管理の基準に関する省令)において、洗浄バリデーションの主たる目的は、製造設備や器具に残留する有効成分、洗浄剤、微生物、エンドトキシン等が、次に製造される製品へ混入し、品質に悪影響を及ぼすリスク(交叉汚染)を確実に低減させることにあります。
具体的には、あらかじめ設定された洗浄手順が、残留物を許容値以下まで一貫して除去できる能力を有することを文書化して検証します。これは、患者様への安全性を確保するための必須要件であり、製造所の品質システムが適切に機能していることを示す証左となります。
医薬品GMPと再生医療(GCTP)における洗浄要件の違い
一般的な低分子医薬品のGMPと比較して、再生医療(GCTP)における洗浄要件にはいくつかの重要な違いがあります。最も大きな特徴は、対象が生きた細胞や組織であるため、最終製品での滅菌が困難であり、製造工程全体を通じた無菌性の確保(アセプティック・プロセッシング)が求められる点です。
また、細胞由来のタンパク質などの汚れは、熱や化学物質による変性を受けやすく、洗浄除去が難しくなるケースがあります。さらに、培地成分や添加剤など、生物由来原料特有のリスク(ウイルス等)も考慮に入れる必要があり、より厳格かつ特殊な洗浄評価が求められます。
汚染管理戦略(CCS)に基づく洗浄プロセスの位置づけ
近年、PIC/S GMP Annex 1の改訂などにより、汚染管理戦略(Contamination Control Strategy: CCS)の重要性が高まっています。洗浄プロセスは、このCCSを構成する重要な要素の一つとして位置づけられます。
単に「洗えばよい」というわけではなく、施設設計、空調管理、更衣手順、そして製造プロセス全体のリスクアセスメントに基づき、洗浄工程がどのように汚染リスクを低減しているかを論理的に説明できなければなりません。つまり、洗浄バリデーションは独立した活動ではなく、包括的な汚染管理の一部として機能させる必要があります。
洗浄バリデーション実施に向けた前提条件と準備
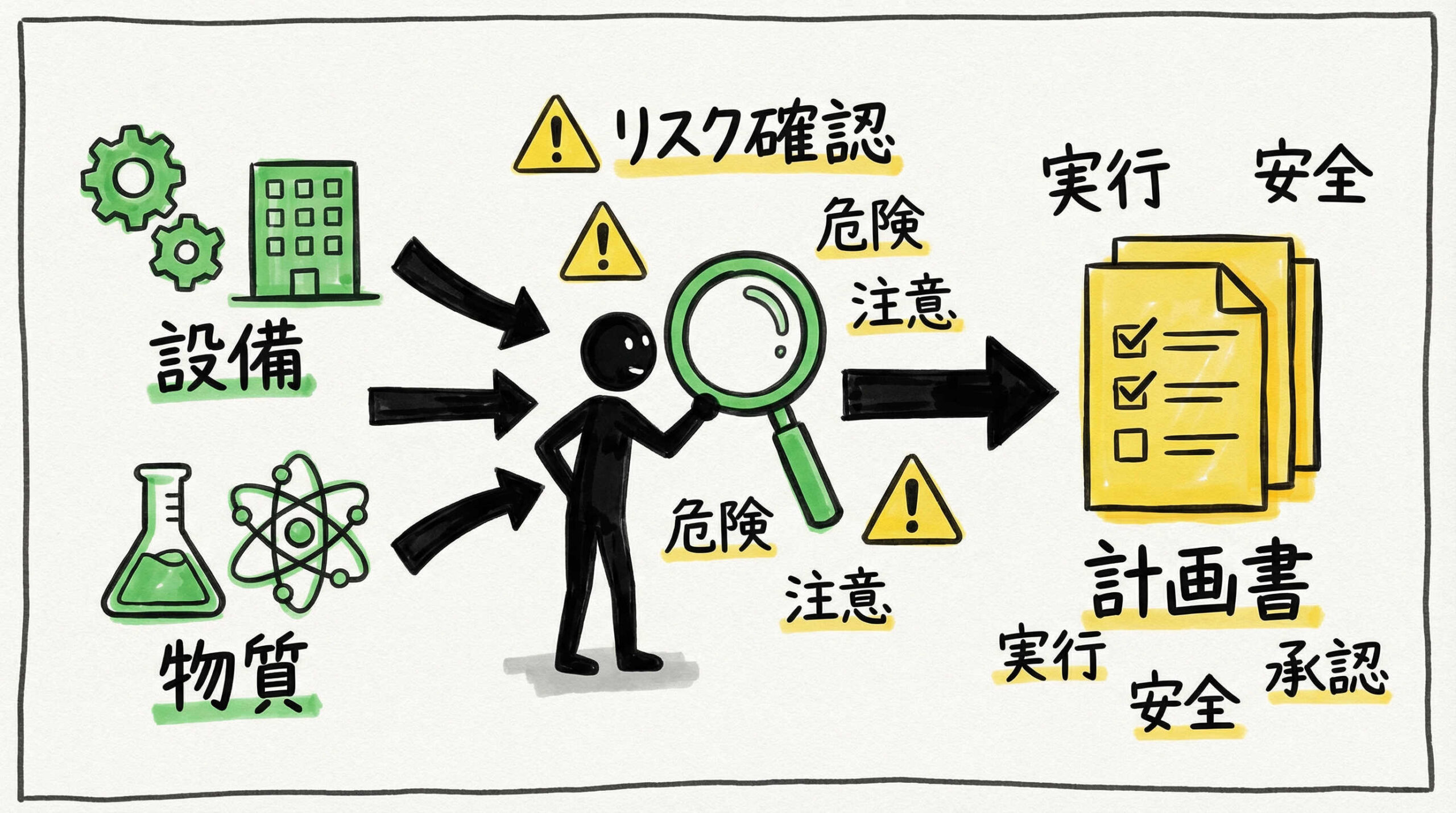
洗浄バリデーションを成功させるためには、実施前の準備段階が非常に重要です。対象となる設備や物質を精査し、リスクに基づいたアプローチを設計することで、効率的かつ効果的なバリデーション計画を立案できます。ここでは、実施要領に盛り込むべき前提条件と準備事項について解説します。
対象設備・機器のグルーピングとワーストケースの選定
製造所内のすべての設備・機器について個別にバリデーションを行うことは、時間とコストの面で現実的ではありません。そこで、構造や材質、洗浄難易度が類似した機器をグループ化し、その中で最も洗浄が困難な条件(ワーストケース)を選定して評価を行う「グルーピング」の手法が有効です。
ワーストケースの選定にあたっては、以下の要素を総合的に評価します。
- 接液部の表面積や材質
- 構造の複雑さ(デッドスペースの有無)
- 汚れの落ちにくさ(溶解性、粘度)
この代表機器でのバリデーション結果をもって、グループ内の他の機器の洗浄性も保証するという論理構成を明確にしておきましょう。
洗浄対象物質(細胞、培地成分、試薬等)の特定
何を除去すべきか、すなわち「洗浄対象物質」を明確に特定することは、分析法の開発や許容値設定の基礎となります。再生医療等製品の場合、以下の成分が主な対象となります。
- 細胞および細胞由来成分: 細胞片、タンパク質、DNAなど
- 培地成分: 糖、アミノ酸、血清成分、成長因子など
- 製造用試薬: 酵素、抗生物質、洗浄剤そのもの
- 分解生成物: プロセス中で発生する可能性のある分解物
これらの物質の物理化学的性質(溶解度、pH安定性など)を把握し、洗浄剤の選定や洗浄パラメータの設定に反映させることが重要です。
ダーティホールドタイム(DHT)とクリーンホールドタイム(CHT)の設定
洗浄プロセスの堅牢性を評価するために、以下の2つの時間制限を設定し、検証する必要があります。
- ダーティホールドタイム(DHT): 製造終了から洗浄開始までの許容時間。汚れが乾燥して固着したり、微生物が増殖したりするリスクを考慮し、最長時間を設定してバリデーションを行います。
- クリーンホールドタイム(CHT): 洗浄・滅菌終了から次の製造使用開始までの有効期限。保管中の再汚染や無菌性の維持を確認します。
これらのタイムリミットをSOPに明記し、実製造で遵守できる現実的な時間を設定することが肝要です。
洗浄手順書(SOP)の確立と作業者の教育訓練
バリデーションを実施する前に、洗浄手順書(SOP)が確定されており、作業者がその手順を正確に再現できる状態にある必要があります。SOPには、洗浄剤の濃度、温度、流量、洗浄時間、すすぎ水量などの重要パラメータを具体的に記載します。
また、特に手洗浄(マニュアル洗浄)を含む場合は、作業者間のバラツキが出やすいため、徹底した教育訓練と実技評価が不可欠です。「誰が洗っても同じ結果になる」レベルまで標準化されて初めて、バリデーションを実施する意義が生まれます。
残留許容値(限度値)の科学的な設定根拠
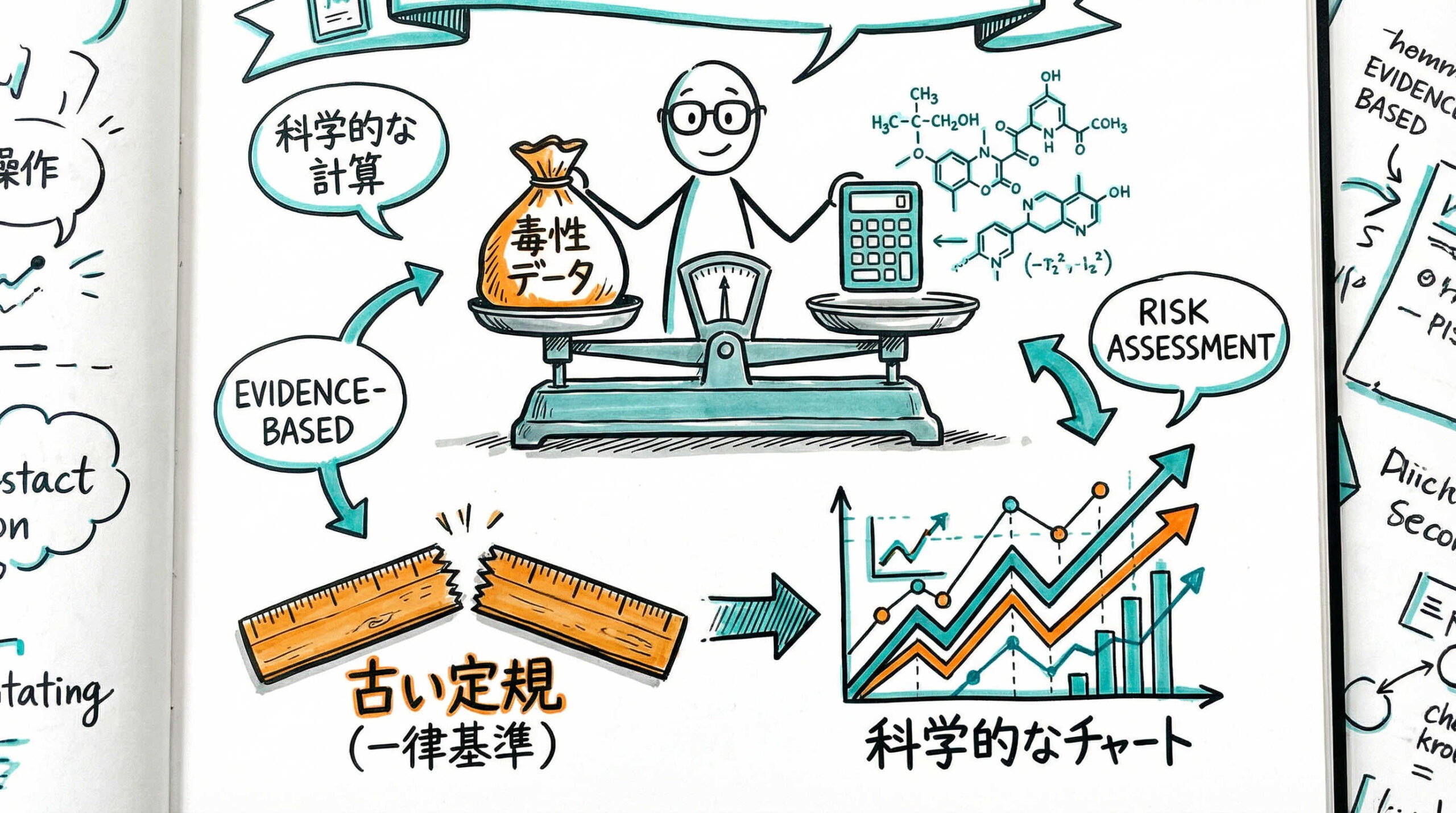
洗浄バリデーションにおいて最も問われるのが「残留許容値(限度値)の設定根拠」です。かつて主流であった一律の基準から、現在は科学的な毒性評価に基づく設定へとシフトしています。ここでは、査察でも重視される許容値設定の考え方と計算手法について詳しく解説します。
毒性学的評価に基づく基準(ADE/PDE値)の適用
現在、国際的な規制調和(ICH Q3シリーズ等)の流れの中で、残留許容値の設定は毒性学的評価に基づくことが標準となっています。具体的には、PDE(Permitted Daily Exposure:一日許容曝露量)やADE(Acceptable Daily Exposure:許容一日曝露量)といった値を算出します。
これは、「その物質を一生涯にわたって毎日摂取しても、健康に悪影響を及ぼさない量」を示すものです。NOAEL(無毒性量)等の毒性データに不確実性係数を適用して算出されます。再生医療等製品においても、使用する試薬や添加剤について、MSDSや文献情報を基にこれらの値を設定し、科学的な安全性を担保する必要があります。
1/1000法および10ppm法の適用可否と限界
従来、残留許容値の設定には「前製品の投与量の1/1000」や「10ppm(100万分の10)」といった基準が広く用いられてきました。これらは簡便で分かりやすい指標ですが、物質固有の毒性を反映していないという科学的な弱点があります。
現在のガイドラインでは、これらの基準のみに依存することは推奨されていません。ただし、毒性データが不足している場合や、ADE/PDE値に基づく計算結果が著しく高く(緩く)なる場合の「歯止め(上限値)」として、これらの従来基準を併用することは依然として有効なアプローチです。複数の基準で計算し、最も厳しい値を採用するのが安全側の判断と言えるでしょう。
微生物およびエンドトキシンの許容基準設定
再生医療等製品は無菌製剤であることが多いため、化学的な残留物だけでなく、微生物およびエンドトキシンの管理が極めて重要です。
- 微生物: 洗浄後の表面におけるバイオバーデン(微生物数)の上限値を設定します。通常は、滅菌前の管理基準などを参考に設定されます。
- エンドトキシン: 特に静脈内投与される製品では、発熱性物質であるエンドトキシンの残留は許されません。注射用水(WFI)の規格などを参考に、厳格な限度値を設定する必要があります。
これらは洗浄工程だけでなく、乾燥や保管工程の管理状態も反映するため、総合的な評価が必要です。
目視確認による残留性評価の有効性と基準
目視確認は、洗浄バリデーションにおいて基本的かつ重要な評価項目です。「目に見える汚れが残っていてはならない」という基準は、いかなる計算上の許容値よりも優先されるべき大前提です。
ただし、目視確認を「定量的」な評価として採用するには、その検出限界を科学的に証明する必要があります。単に「きれいです」と報告するだけでは不十分であり、照明条件、観察距離、角度、観察者の視力などを規定した上で、その有効性を裏付けるデータが求められます。
スパイク回収率試験における回収率の許容範囲
目視確認の信頼性を担保するために実施されるのが「スパイク回収率試験」に類似した目視限界試験です。これは、実際の機器材質と同じテストピース(クーポン)に、既知量の汚れを塗布し、どの程度の濃度までなら目視で検出できるかを確認するものです。
一般的に、熟練した作業者が適切な条件下で観察した場合、数µg/cm²程度の汚れを検出できるとされています。この試験を行い、「目視で汚れが見えない状態であれば、残留量は許容値以下である」という相関関係を証明することで、日常的な洗浄確認における目視検査の妥当性を主張できます。
具体的な洗浄バリデーションの実施手順フロー
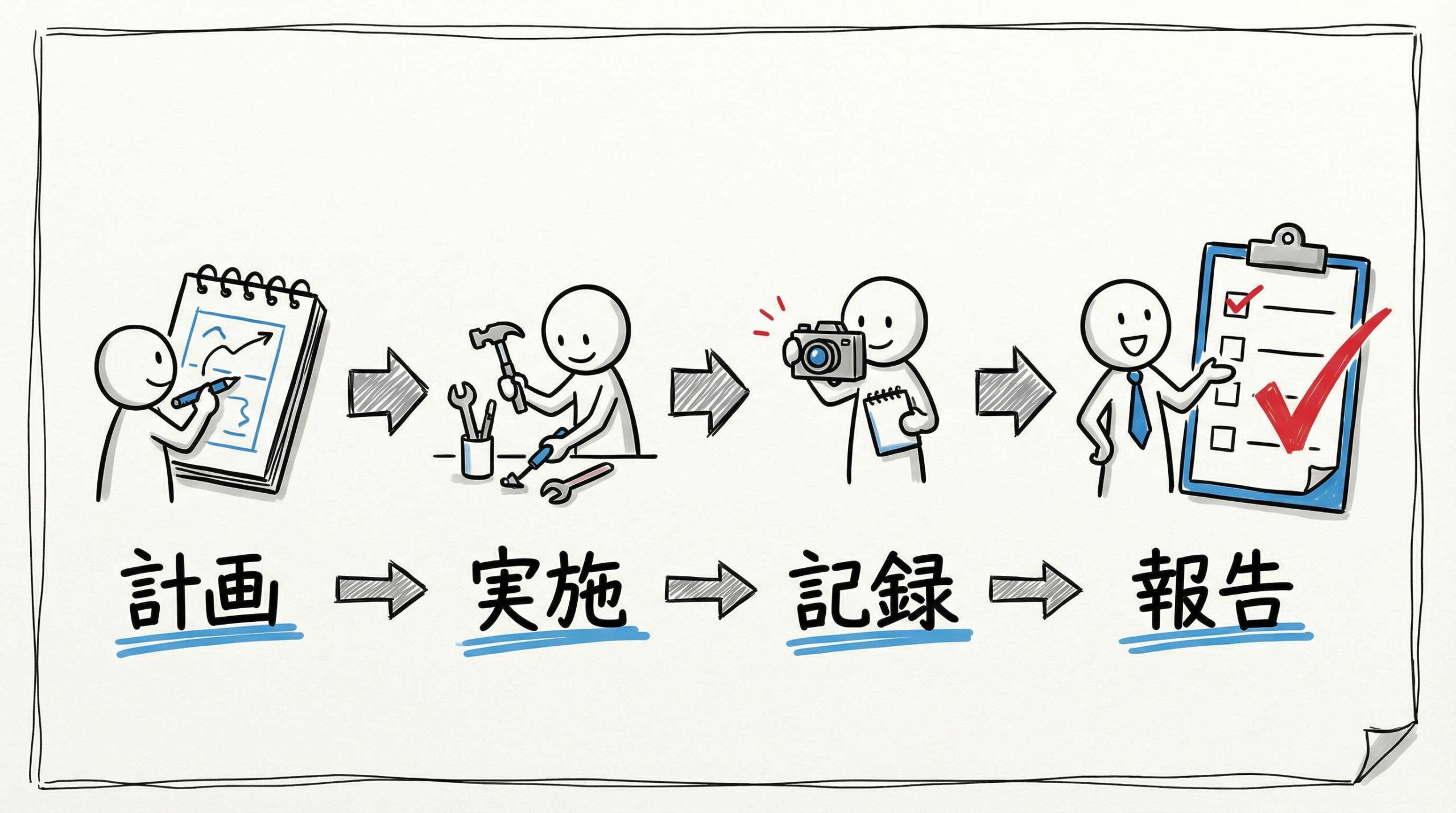
理論的な準備が整ったら、いよいよ実際のバリデーション作業に移ります。計画から報告書の作成まで、一連の流れをスムーズに進めるための具体的なステップを確認しましょう。各段階での確実な実施と記録が、後の査察対応での信頼性につながります。
ステップ1:バリデーション実施計画書(プロトコル)の作成
最初のステップは、バリデーション実施計画書(プロトコル)の作成です。ここには、目的、範囲、責任体制、対象機器、洗浄手順、サンプリング計画、分析方法、判定基準(許容値)などを詳細に記述します。
計画書は、実施前に品質保証部門(QA)等の承認を得る必要があります。計画段階で不備があると、得られたデータの信頼性が損なわれるため、リスク評価に基づいた論理的な計画になっているか、入念にレビューを行いましょう。
ステップ2:実生産規模での洗浄実施(3ロットの実績)
承認された計画書に基づき、実際の製造設備を用いて洗浄操作を実施します。バリデーションの信頼性を確保するため、通常は連続する3ロット(または3回の洗浄操作)で実施し、結果の再現性を確認します(3回繰り返し原則)。
この際、SOPに記載された手順を逸脱することなく、忠実に作業を行うことが重要です。また、DHT(汚染保持時間)などのワーストケース条件を計画に組み込んでいる場合は、その条件通りに実施されているか厳密に管理します。
ステップ3:サンプリングの実施(スワブ法・リンス法)
洗浄終了後、あらかじめ設定した箇所からサンプリングを行います。サンプリング箇所は、洗浄しにくい場所(デッドレグ、バルブ周辺、撹拌翼の裏側など)をリスク評価に基づいて選定します。
手法としては、物理的に汚れを拭き取る「スワブ法」と、溶媒ですすぎ洗いをして回収する「リンス法」を適切に組み合わせます。サンプリング操作自体が汚染の原因とならないよう、無菌操作や適切な器具の使用に留意してください。
ステップ4:検体の分析とデータ評価
回収した検体を、バリデーションされた分析法(TOC、HPLC等)を用いて分析します。ここで得られた定量値から、機器表面全体の残留量を換算し、事前に設定した許容値と比較します。
分析データの評価においては、回収率(Recovery Rate)の補正を忘れないようにしましょう。サンプリング操作で100%の汚れが回収できるわけではないため、事前の回収率試験で得られた係数を用いて、実際の残留量を推定する必要があります。
ステップ5:バリデーション報告書の作成と承認
全てのデータが出揃ったら、結果を総括し、バリデーション報告書を作成します。各ロットの結果、逸脱の有無、判定基準への適合状況を記載し、最終的に「当該洗浄手順は妥当である」という結論を導き出します。
もし判定基準を満たせなかった場合は、原因を究明し、洗浄手順の見直しや再バリデーションの必要性を検討します。報告書は責任者の承認を経て保管され、将来の査察時の重要な証拠書類となります。
サンプリング方法と分析手法の選定ポイント
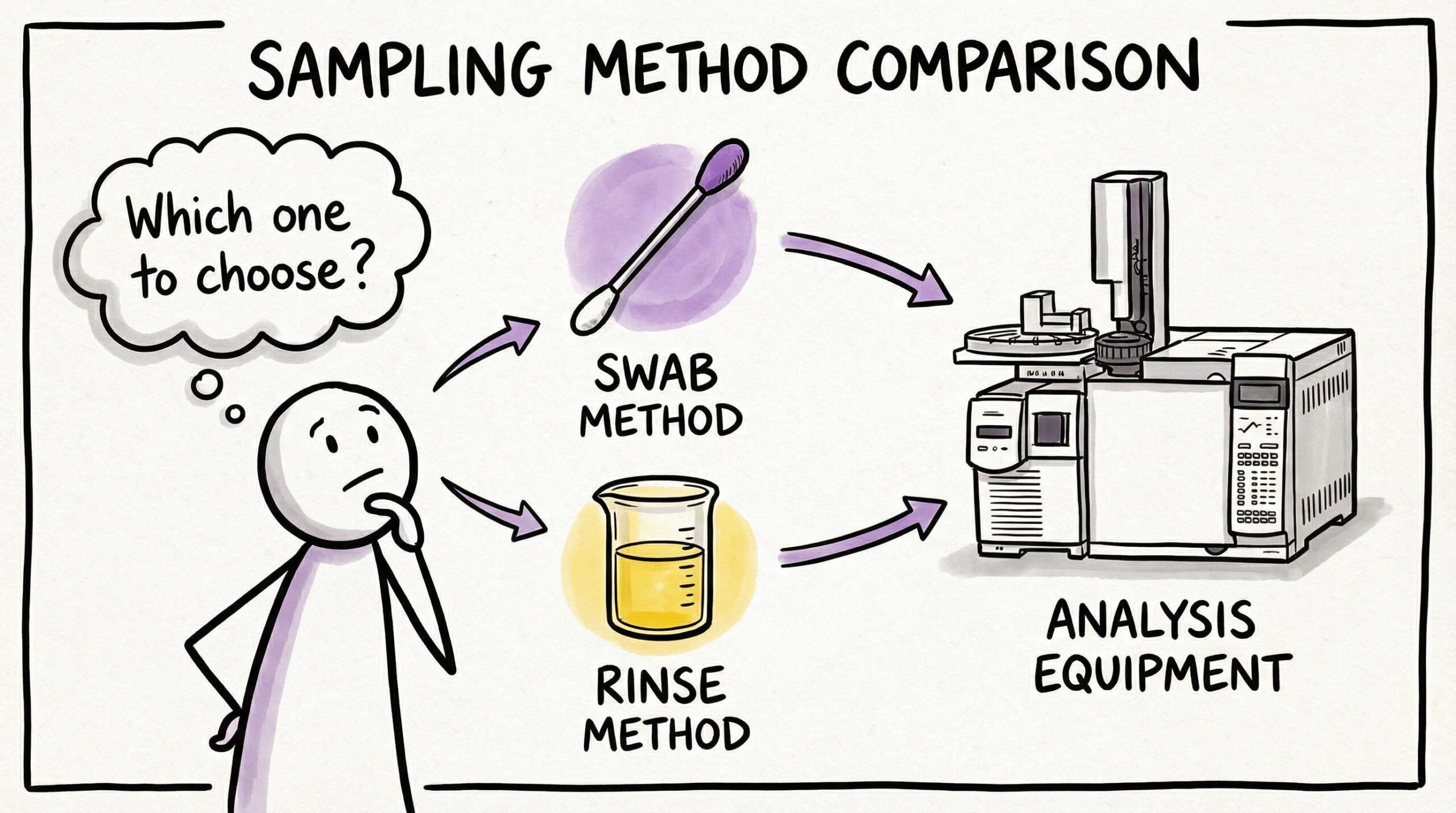
正しい評価結果を得るためには、適切なサンプリング方法と分析手法の選定が欠かせません。対象機器の形状や残留物の性質に応じて、最適な組み合わせを選択するポイントを解説します。データの信頼性を左右する重要な技術的要素です。
スワブ法(ふき取り法)の適用箇所と実施手順
スワブ法は、特定の面積(通常25cm²や100cm²)を専用のスワブ材で規定回数拭き取る方法です。物理的な力を加えて汚れを回収するため、乾燥・固着した汚れに対して有効であり、特定の場所(ホットスポット)を直接評価できる利点があります。
適用箇所としては、手が届く範囲の平滑な表面、分解可能な部品、洗浄難易度が高いと特定された箇所が適しています。実施手順(拭き取り方向、回数、圧力など)を標準化し、作業者によるバラツキを抑えることが重要です。
リンス法(すすぎ法)の適用条件と注意点
リンス法は、機器全体または配管内部などに溶媒を流し、その回収液を分析する方法です。スワブ法では届かない配管内部や複雑な形状の機器に適しており、接液部全体(広範囲)を評価できる利点があります。
ただし、汚れが溶媒に溶解することが前提条件となります。また、残留物が大量の溶媒で希釈されるため、検出感度が低下する可能性があります。リンス法を採用する場合は、溶媒が機器の隅々まで行き渡っているか(乱流の確保など)を確認する必要があります。
ダイレクトサンプリング法の活用
近年、表面の残留物を直接測定する技術(ダイレクトサンプリング)の活用も進んでいます。例えば、FT-IR(フーリエ変換赤外分光光度計)やラマン分光法を用いたポータブル機器などが挙げられます。
これらはサンプリング操作による誤差を排除でき、リアルタイムでの測定が可能というメリットがあります。ただし、機器の導入コストや、曲面・狭所での測定限界などの課題もあるため、従来法との相関性を確認した上で、補助的なツールとして活用するのが一般的です。
特異的分析法と非特異的分析法(TOC等)の使い分け
分析手法には、特定の成分のみを検出する「特異的分析法」(HPLC、ELISA等)と、有機物全体を検出する「非特異的分析法」(TOC:全有機体炭素計等)があります。
再生医療分野では、培地成分や細胞由来成分など多種多様な有機物が混在するため、TOC法が広く採用されています。TOC法は全ての有機炭素を検出できるため、予期せぬ汚染物質も含めたワーストケース評価として有用です。一方、特定の毒性が高い物質(薬剤等)を厳密に管理したい場合は、特異的分析法を選択します。
再生医療等製品特有の課題と対策
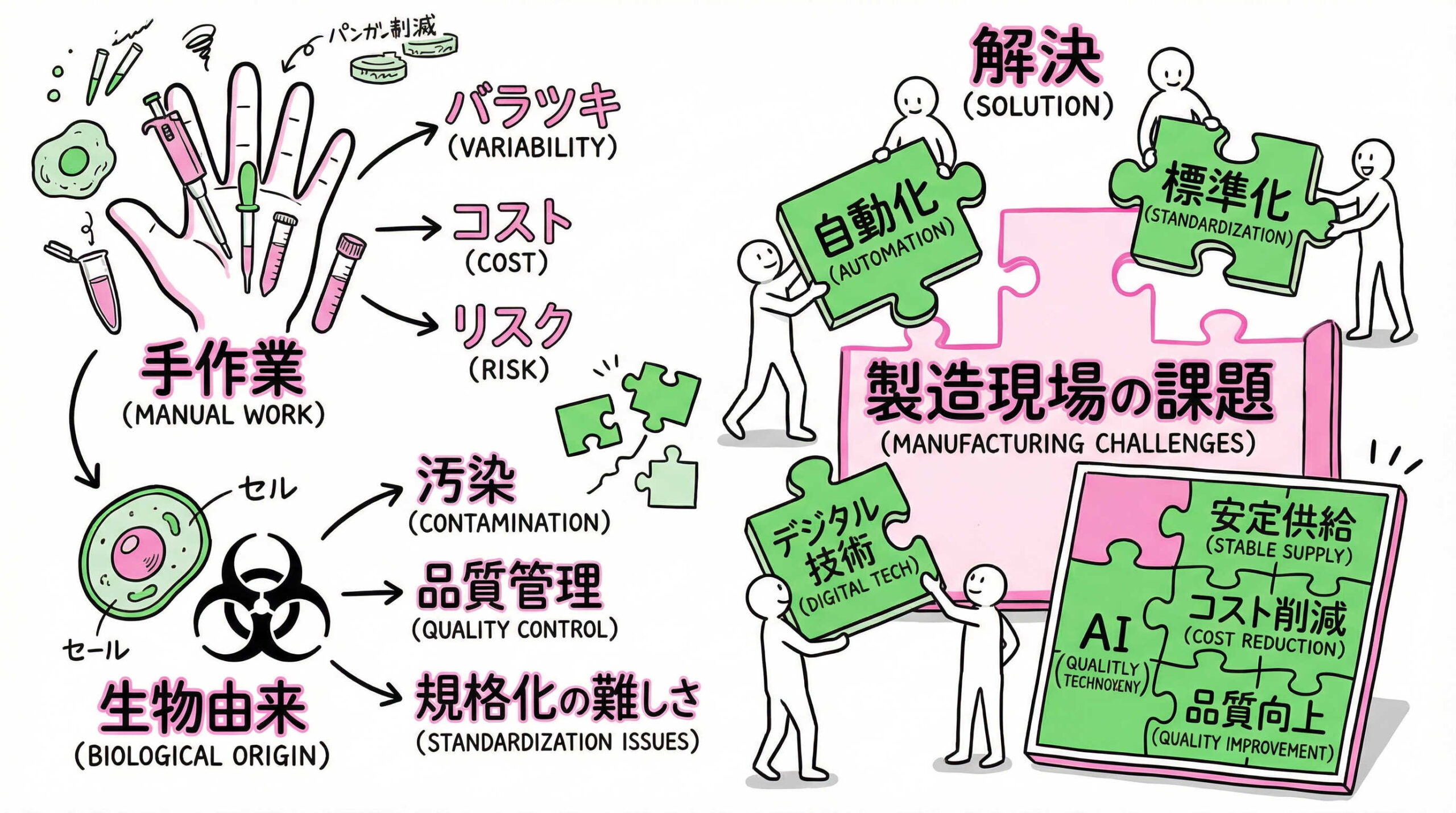
再生医療等製品の製造には、従来の医薬品製造とは異なる特有の難しさがあります。手作業への依存度の高さや、生物由来原料のリスクなど、現場が直面しやすい課題とその対策について掘り下げていきます。
手洗浄(マニュアル洗浄)における再現性の確保
再生医療の製造現場では、小規模かつ複雑な器具を使用することが多く、CIP(定置洗浄)よりもマニュアル洗浄(手洗浄)が多用されます。手洗浄の最大の課題は、作業者による「再現性の確保」です。
対策としては、SOPの細分化(ブラシの種類、こする回数、水温等の指定)に加え、動画マニュアルの活用や、定期的な技量認定制度の導入が有効です。また、洗浄後の目視確認をダブルチェック体制にするなど、ヒューマンエラーを検出する仕組みも重要です。
専用設備と共用設備におけるバリデーションアプローチの違い
交叉汚染防止の観点からは、製品ごとに専用設備(シングルユース製品含む)を使用するのが理想的です。しかし、コストやスペースの制約から共用設備を使用せざるを得ない場合もあります。
共用設備の場合は、前製品から次製品へのキャリーオーバーを厳密に評価する洗浄バリデーションが必須となります。一方、専用設備であっても、ロット間の洗浄(同一製品の繰り返し製造)に関するバリデーションは必要です。ただし、共用設備に比べてリスクは低いため、評価項目を簡素化できる場合があります。
ウイルス不活化・除去工程を含む洗浄評価
ヒトや動物由来の原料を使用する場合、ウイルス汚染のリスクは無視できません。製造工程にウイルス不活化・除去工程が含まれている場合、その工程後の洗浄プロセスが、除去されたウイルスを再汚染させないことを確認する必要があります。
洗浄バリデーションにおいては、ウイルスそのものを測定することは困難なため、モデルウイルスを用いたクリアランス試験のデータを参照したり、適切な洗浄剤(酸、アルカリ等)の使用による不活化効果を理論的に説明したりするアプローチが取られます。
キャンペーン生産における洗浄評価(洗浄ベリフィケーション)
同一製品を連続して製造する「キャンペーン生産」では、ロットごとの完全洗浄を行わず、簡易洗浄(マイナー洗浄)で済ませる場合があります。この場合、キャンペーン終了後の完全洗浄(メジャー洗浄)までの間に、汚れが蓄積して許容値を超えないことを確認する必要があります。
ここでは、定期的なモニタリングや、製品品質データの確認を通じて、洗浄状態が維持されていることを継続的に検証する「洗浄ベリフィケーション」の考え方が重要になります。キャンペーンの最大ロット数を設定する根拠としても用いられます。
洗浄バリデーションの維持管理と再バリデーション
バリデーションは「一度やれば終わり」ではありません。製造プロセスが続く限り、洗浄工程が常に管理された状態(Validated State)にあることを維持し続ける必要があります。ここでは、日常の管理手法と再バリデーションのタイミングについて解説します。
定期的なモニタリングとトレンド分析
バリデーション完了後も、日常的な洗浄記録の確認や、定期的なサンプリング(TOC測定や導電率測定など)を通じて、洗浄プロセスが安定していることを監視します。これを「継続的プロセス検証」とも呼びます。
得られたデータをトレンド分析し、数値が徐々に上昇している傾向(ドリフト)が見られた場合は、許容値内であっても早期に対策を講じます。これにより、突発的な逸脱を防ぎ、恒常的な品質を保証することができます。
変更管理(チェンジコントロール)時の再評価基準
製造設備、洗浄剤、洗浄手順、製造プロセス(原料変更など)に変更が生じる場合は、変更管理(チェンジコントロール)の手続きに則り、洗浄バリデーションへの影響を評価します。
軽微な変更であれば机上の評価(アセスメント)で済む場合もありますが、洗浄効果に影響を与える可能性がある重要な変更の場合は、再バリデーション(リバリデーション)の実施が必要です。変更を実施する前に評価を行うことが原則です。
逸脱発生時の対応プロセスと是正措置
万が一、洗浄後の検査で不合格となったり、手順からの逸脱が発生したりした場合は、直ちに逸脱処理プロセスを開始します。単に再洗浄して良しとするのではなく、「なぜ逸脱が起きたのか」という根本原因(Root Cause)を究明しなければなりません。
原因が手順の不備にあるのか、設備の劣化なのか、作業者のミスなのかを特定し、CAPA(是正措置・予防措置)を策定・実施します。このプロセスが適切に回っていることが、GMP/GCTP体制の健全性を示します。
まとめ
再生医療における洗浄バリデーションは、患者様の安全を守るための「最後の砦」の一つです。GCTP省令や関連ガイドラインに基づき、科学的な根拠(毒性評価やリスクアセスメント)を持った実施要領を策定することが求められます。
本記事で解説した通り、適切なグルーピング、対象物質の特定、そして堅実なサンプリングと分析の実施は、信頼性の高いバリデーションの鍵となります。また、一度確立したプロセスも、変更管理やモニタリングを通じて維持し続ける姿勢が不可欠です。確実な洗浄バリデーションの実践により、高品質な再生医療等製品の安定供給を目指しましょう。
洗浄バリデーションの実施要領についてよくある質問

以下に、洗浄バリデーションの実施要領に関して、現場担当者からよく寄せられる質問をまとめました。実務の参考としてご活用ください。
- Q1. 洗浄バリデーションはどのタイミングで実施すべきですか?
- A1. 新規設備の導入時、製造プロセスの変更時、洗浄手順や洗浄剤の変更時に実施する必要があります。また、定期的(例えば年1回)な再評価も推奨されます。
- Q2. 専用設備(シングルユース)の場合でも洗浄バリデーションは必要ですか?
- A2. シングルユース部材は洗浄不要ですが、コネクタやハウジングなど再使用する部分がある場合は必要です。また、専用設備でもロット間の洗浄評価は必要となります。
- Q3. 毒性データがない新規物質の残留許容値はどう設定すればよいですか?
- A3. 類似物質の毒性データを参照するか、TTC(毒性学的懸念の閾値)アプローチを用いて設定することを検討します。専門家による毒性評価を受けることが望ましいでしょう。
- Q4. 洗浄剤の残留確認はどのように行いますか?
- A4. 洗浄剤の成分(界面活性剤など)を指標とし、TOCや導電率、または特異的な分析法を用いて残留量が許容値以下であることを確認します。メーカーから開示されるデータも参考にします。
- Q5. バリデーション実施時の「3ロット」は必須ですか?
- A5. 一般的に再現性を確認するために3ロット(3回の繰り返し)が求められます。ただし、科学的な正当性があれば、リスクベースアプローチにより回数を調整できる場合もあります。
<script type="application/ld+json">
{
"@context": "https://schema.org",
"@type": "FAQPage",
"mainEntity": [
{
"@type": "Question",
"name": "洗浄バリデーションはどのタイミングで実施すべきですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "新規設備の導入時、製造プロセスの変更時、洗浄手順や洗浄剤の変更時に実施する必要があります。また、定期的(例えば年1回)な再評価も推奨されます。"
}
},
{
"@type": "Question",
"name": "専用設備(シングルユース)の場合でも洗浄バリデーションは必要ですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "シングルユース部材は洗浄不要ですが、コネクタやハウジングなど再使用する部分がある場合は必要です。また、専用設備でもロット間の洗浄評価は必要となります。"
}
},
{
"@type": "Question",
"name": "毒性データがない新規物質の残留許容値はどう設定すればよいですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "類似物質の毒性データを参照するか、TTC(毒性学的懸念の閾値)アプローチを用いて設定することを検討します。専門家による毒性評価を受けることが望ましいでしょう。"
}
},
{
"@type": "Question",
"name": "洗浄剤の残留確認はどのように行いますか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "洗浄剤の成分(界面活性剤など)を指標とし、TOCや導電率、または特異的な分析法を用いて残留量が許容値以下であることを確認します。メーカーから開示されるデータも参考にします。"
}
},
{
"@type": "Question",
"name": "バリデーション実施時の「3ロット」は必須ですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "一般的に再現性を確認するために3ロット(3回の繰り返し)が求められます。ただし、科学的な正当性があれば、リスクベースアプローチにより回数を調整できる場合もあります。"
}
}
]
}
</script>