
再生医療等製品の製造において、施設の品質管理は製品の安全性と有効性を担保する上で極めて重要な役割を果たします。特に新規に細胞培養加工施設(CPC)を立ち上げる際や、大規模な改修を行う際には、「施設バリデーション」の計画と実施が避けて通れない課題となります。しかし、DQ、IQ、OQ、PQといった適格性評価の流れや、それぞれの具体的な実施内容について、体系的に整理することは容易ではありません。
本記事では、再生医療分野における施設バリデーションの全体像を、GCTP省令やVモデルの概念を交えながら分かりやすく解説します。各適格性評価のポイントから、主要設備の検証項目、そしてバリデーション完了後の維持管理に至るまで、実務に即した情報をお届けします。貴施設のバリデーション計画策定の一助となれば幸いです。
施設バリデーションの全体像とは?DQ/IQ/OQ/PQの役割と流れ
施設バリデーションは、単に設備が動くことを確認するだけでなく、期待される品質を恒常的に保証できることを科学的に検証し、文書化するプロセスです。ここではまず、その基本概念と全体的な流れについて、適格性評価(DQ/IQ/OQ/PQ)を中心に解説します。
施設バリデーションの定義と再生医療における重要性
バリデーションとは、製造所の構造設備や手順、工程などが期待される結果を与えることを検証し、これを文書化することです。特に再生医療においては、最終製品の無菌試験やすべての品質試験を行うことが難しいケースが多く、製造プロセス全体での品質保証が求められます。
施設バリデーションは、その基盤となるハードウェア(CPCや製造設備)が、意図した通りに機能し、清浄度などの環境条件を維持できることを証明するものです。これが不十分であれば、どれほど優れた製造手順があっても、製品の品質を担保することはできません。つまり、施設バリデーションは「品質を作り込む」ための土台作りと言えるでしょう。
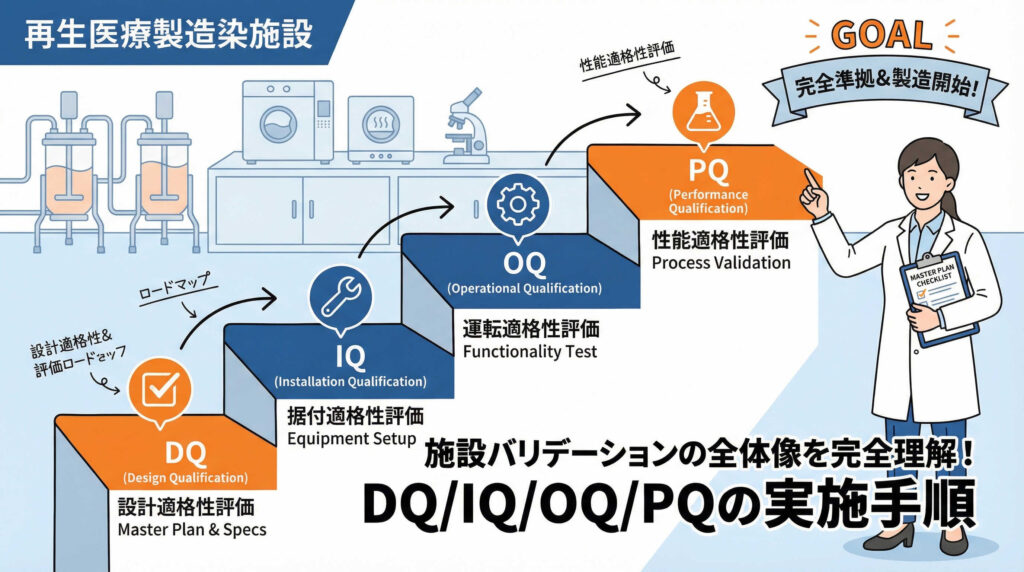
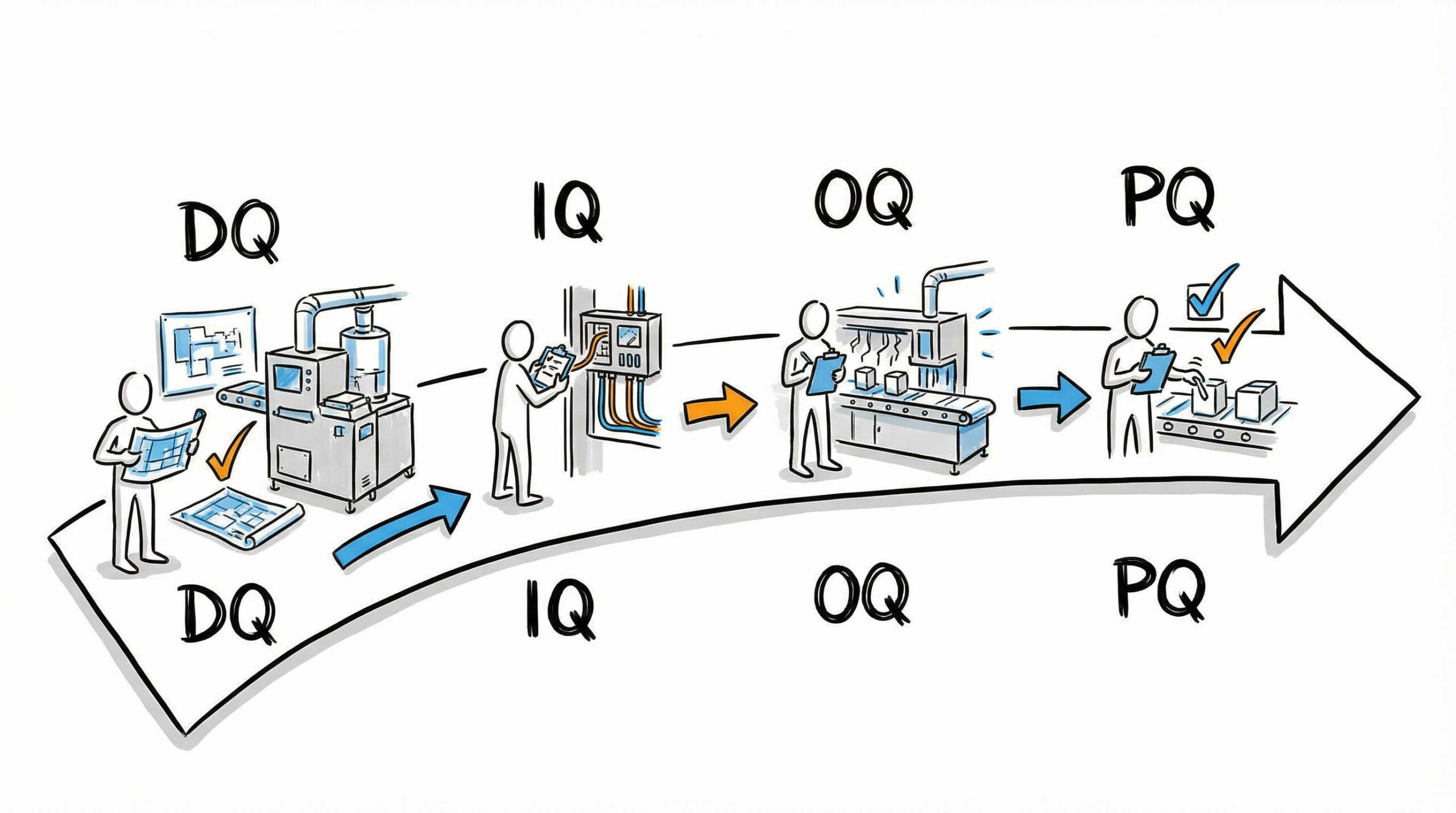
適格性評価(DQ/IQ/OQ/PQ)のVモデルによる相互関係
適格性評価は、一般的に「Vモデル」と呼ばれる概念に基づいて進められます。Vモデルは、設計段階から検証段階までのプロセスをV字型に配置し、左右の対応関係を示したものです。
- DQ(設計時適格性評価): ユーザー要求仕様書(URS)に基づき、設計が適切かを確認します。
- IQ(据付時適格性評価): 機器が設計通りに設置されたかを確認します。
- OQ(運転時適格性評価): 機器が運転範囲内で正常に動作するかを確認します。
- PQ(性能適格性評価): 実際の製造条件に近い負荷状態で性能を確認します。
このように、上流の設計から下流の性能確認へと段階的に検証を進めることで、抜け漏れのない品質保証が可能となります。
GCTP省令に基づいたバリデーションの法的要件
再生医療等製品の製造管理及び品質管理の基準に関する省令(GCTP省令)では、構造設備が適切に設計、配置、設置され、かつ維持管理されることが求められています。バリデーションは、この法的要件を満たすための必須事項です。
具体的には、GCTP省令において、バリデーション指針の作成や責任者の設置、実施計画書および報告書の作成が規定されています。当局の査察においても、バリデーション記録は必ず確認される重要書類です。したがって、法令遵守(コンプライアンス)の観点からも、科学的根拠に基づいたバリデーションの実施と記録保管は不可欠な業務となります。
【段階別詳細】各適格性評価の実施内容と判定基準
適格性評価は、DQからPQへと順を追って実施することが原則です。各段階で何を確認し、どのような基準で判定するのか、その詳細を見ていきましょう。前の段階が完了し、承認されて初めて次の段階へ進むことができます。
DQ(設計時適格性評価):要求仕様(URS)と設計の整合性確認
DQ(Design Qualification)は、設備の導入や建設の最も初期段階に行う評価です。ここでは、作成された設計図書や仕様書が、ユーザー要求仕様書(URS)およびGMP/GCTP要件を満たしているかを机上で確認します。
具体的には、以下のような項目を照合します。
- 空調システム: 清浄度クラス、室圧設定、温湿度条件が要求通りか。
- 動線計画: 人や物の動きが交差汚染を防ぐ設計になっているか。
- 材質: 清掃や消毒に耐えうる材質が選定されているか。
この段階で不備を見逃すと、後の修正に多大なコストと時間がかかるため、慎重な確認が必要です。
IQ(据付時適格性評価):機器・設備の設置状況と仕様の確認
IQ(Installation Qualification)は、設備や機器が納入され、設置された直後に行う評価です。設計図や仕様書通りに正しく据え付けられ、ユーティリティ(電気、水、ガスなど)が適切に接続されているかを確認します。
主な確認項目は以下の通りです。
- 現物確認: 型式、シリアル番号、材質が仕様書と一致しているか。
- 設置状況: 水平度、配管・配線の接続、設置環境(スペース等)。
- 校正: 計器類が校正されており、証明書が揃っているか。
IQでは、機器そのものの仕様確認に加え、付属品やマニュアルの有無など、物理的な側面を網羅的にチェックします。
OQ(運転時適格性評価):運転範囲と機能性能の検証
OQ(Operational Qualification)は、機器が規定された運転範囲内で正常に動作することを確認する評価です。通常は無負荷の状態(製品や検体を入れない状態)で実施します。
検証内容には以下が含まれます。
- 機能試験: スイッチ操作、タッチパネルの反応、各種モードの動作確認。
- 警報試験: 設定値を外れた際にアラームが鳴るか、インターロックが作動するか。
- 停電復帰試験: 電源遮断後の再起動が適切に行われるか。
OQによって、機器単体としての機能的性能が保証されます。ここで重要なのは、最悪条件(ワーストケース)を含めた範囲での動作確認を行うことです。
PQ(性能適格性評価):実負荷状態での性能とプロセスの安定性
PQ(Performance Qualification)は、バリデーションの最終段階であり、実際の製造環境に近い状態、あるいは負荷をかけた状態で性能を検証します。
再生医療施設におけるPQの例:
- 空調システム: 人が入室し作業している状態での室圧、温度、清浄度維持。
- 培養装置: 実際に培地等を入れた状態での温度分布やガス濃度安定性。
PQでは、プロセス全体が安定して稼働し、製品品質に影響を与えるパラメータが許容範囲内に収まり続けることを実証します。このデータが、将来的な日常管理基準の根拠となります。
再生医療施設(CPC)でバリデーション対象となる主要設備・機器
再生医療施設(CPC)には、細胞の品質を守るために高度に制御された設備が必要です。ここでは、特にバリデーションの重要性が高い主要な設備・機器について、具体的な検証ポイントを解説します。
空調システム(HVAC):室圧制御と清浄度維持の検証
空調システム(HVAC)は、CPC内の清浄度を維持し、交差汚染を防ぐための要となる設備です。バリデーションでは、HEPAフィルターの完全性試験(リーク試験)を行い、粒子が除去されていることを確認します。
また、各部屋間の室圧制御(差圧)も重要です。清浄度の高い部屋から低い部屋へ気流が流れているか、ドアの開閉時に室圧がどのように変動し、どれくらいの時間で復帰するかを検証します。さらに、温湿度が細胞培養に適した範囲で制御されているかも、長時間のモニタリングを通じて確認します。
安全キャビネット(BSC):気流速度と封じ込め性能の確認
安全キャビネット(BSC)は、無菌操作を行うための局所的な清浄空間を提供し、同時に作業者を暴露から守る役割があります。
バリデーションにおける主な確認項目:
- 気流速度: 吸込み風速と吹出し風速が規定範囲内か。
- 気流可視化(スモークテスト): 内部の気流が乱れず、外部への漏洩や外部からの流入がないか。
- 清浄度: 作業エリア内がグレードA相当を維持しているか。
これらの試験により、封じ込め性能と無菌環境の両立を証明します。
CO2インキュベーター:庫内環境の均一性と復帰性能
CO2インキュベーターは、細胞が増殖するための環境(温度、CO2濃度、湿度)を直接提供する機器であり、製品品質に直結します。
ここでは、庫内の温度分布とCO2濃度分布の均一性が重要です。複数のポイントにセンサーを設置し、場所によるばらつきがないかを確認します。また、扉を開閉した後の温度・濃度復帰時間も測定し、作業時の環境変動が許容範囲内であることを検証します。停電時の温度保持能力なども、リスク管理の観点から確認すべき項目です。
細胞保存用機器(ディープフリーザー・液体窒素タンク)の温度分布
製造された細胞製品や原料細胞を保管するディープフリーザーや液体窒素タンクも、バリデーションの対象です。保管温度の逸脱は、細胞の生存率や機能に致命的な影響を与える可能性があります。
PQでは、庫内にサンプル(ダミー)を充填した状態で、庫内各所の温度分布を測定します(温度マッピング)。また、扉の開閉に伴う温度上昇の程度や、電源喪失時の温度上昇カーブを確認し、緊急時の対応時間を算出する根拠とします。液体窒素タンクの場合は、液面レベルの監視機能や自動供給機能の検証も含まれます。
施設バリデーションを成功させるための実施手順とドキュメント管理
バリデーションは、実施する行為そのものと同様に、それを計画し記録するドキュメントワークが極めて重要です。適切な手順と文書管理がなければ、バリデーションは成立しません。成功のための実務フローを見ていきましょう。
バリデーションマスタープラン(VMP)の策定と位置づけ
バリデーション活動の全体像を定義する最上位文書が、バリデーションマスタープラン(VMP)です。VMPには、バリデーションの方針、対象範囲、組織体制、スケジュール、参照規格などを明記します。
個別の機器に対するIQ/OQ/PQを実施する前に、まずこのVMPを策定し、施設全体としてどのような戦略で品質保証を行うかを明確にする必要があります。これにより、プロジェクト関係者間での認識のズレを防ぎ、効率的かつ整合性の取れたバリデーション活動が可能となります。
リスクアセスメント手法を用いた重要管理項目の抽出
すべての項目を均一に検証することは、時間とコストの観点から現実的ではありません。そこで重要になるのがリスクアセスメントです。製品品質への影響度と発生頻度などを分析し、重要管理項目(CQA/CPP)を特定します。
例えば、FMEA(故障モード影響解析)などの手法を用いて、「どの機能が故障すると無菌性が損なわれるか」を評価します。リスクが高いと判断された項目については、バリデーションの試験項目を厚くし、判定基準を厳格にするなど、メリハリのある計画を立てることが推奨されます。
バリデーション計画書・報告書の作成フローと承認
実際のバリデーションは、以下のフローで文書化しながら進めます。
- 計画書作成: 実施内容、方法、判定基準を記載した計画書を作成し、品質保証責任者が承認します。
- 実施と記録: 計画書に基づき試験を実施し、生データを含む記録を残します。
- 報告書作成: 結果をまとめ、判定基準を満たしているか評価します。
- 承認: 責任者が報告書を承認し、バリデーション完了となります。
計画書承認前に実施した試験は無効となるため、日付の順序には厳格な注意が必要です。
逸脱発生時の対応手順と是正措置・予防措置(CAPA)
バリデーション実施中に、測定値が判定基準を外れたり、機器の動作不良が起きたりすることを「逸脱」と呼びます。逸脱が発生した場合、単にやり直すのではなく、必ず記録に残し、原因を究明しなければなりません。
原因が手順の誤りなのか、機器の不具合なのかを特定し、是正措置(Corrective Action)を行います。さらに、再発を防ぐための予防措置(Preventive Action)を講じます。これらCAPA(是正・予防措置)のプロセスを経て、再試験を行い、問題が解決されたことを確認して初めてバリデーションは完了します。
バリデーション完了後の維持管理と再バリデーション
バリデーションは一度実施して終わりではありません。施設の稼働後も、その適格性が維持されていることを継続的に確認する必要があります。運用フェーズにおける管理について解説します。
変更管理プロセスと再バリデーションの実施判断
施設の運用中に、設備の改造、部品の交換、設定値の変更などが必要になることがあります。このような場合、その変更が製品品質に悪影響を与えないかを評価する「変更管理」プロセスが必要です。
変更の内容やリスクの大きさに応じて、再バリデーション(リバリデーション)の要否を判断します。例えば、空調機のファンを交換した場合、単なる動作確認で済むのか、室圧や清浄度の再測定まで必要なのかを検討します。この判断プロセスも文書化し、記録として残すことがGCTP省令で求められています。
定期的なバリデーション(定期点検)のスケジュール管理
変更がない場合でも、経年劣化により設備の性能が低下する可能性があります。そのため、あらかじめ定めた期間ごとに「定期バリデーション」や定期点検を実施します。
- 定期的な測定: HEPAフィルターのリーク試験や室圧測定(年1回など)。
- 校正(キャリブレーション): 温度計や圧力計などの計測機器の精度確認。
これらを年間スケジュール(バリデーションスケジュール)に組み込み、計画的に実施することで、施設が常に適格な状態(バリデートされた状態)にあることを保証します。
まとめ

施設バリデーションは、再生医療等製品の品質を根底から支える重要なプロセスです。DQ、IQ、OQ、PQという一連の流れ(Vモデル)を理解し、GCTP省令に基づいた適切な計画と実施を行うことが、安全で有効な製品を患者様に届けるための第一歩となります。
また、バリデーションは「文書に始まり文書に終わる」と言われるほど、ドキュメント管理が重要です。計画書、実施記録、報告書、そして逸脱管理や変更管理といった記録の積み重ねが、貴施設の品質保証体制の信頼性を証明します。
最初は膨大な作業量に圧倒されるかもしれませんが、リスクアセスメントを活用して重要項目を見極め、一つひとつ着実に進めていくことが成功の鍵です。社内リソースだけで対応が難しい場合は、専門家の知見を借りることも有効な選択肢でしょう。確実なバリデーションを実施し、信頼される再生医療施設を築き上げてください。
施設バリデーションの全体像(DQ/IQ/OQ/PQ)についてよくある質問

よくある質問
以下は、施設バリデーション(DQ/IQ/OQ/PQ)に関してよく寄せられる質問とその回答です。
- Q1. 施設バリデーションにはどのくらいの期間が必要ですか?
- 施設の規模や複雑さによりますが、新規CPCの場合、計画策定からPQ完了まで数ヶ月から半年程度かかることが一般的です。DQは設計段階で行い、IQ/OQは施工完了後、PQは運用開始前に行うため、建設スケジュールと密接に連携する必要があります。
- Q2. 既存の施設でバリデーション記録がない場合、どうすればよいですか?
- 「回顧的バリデーション(レトロスペクティブ・バリデーション)」という手法がありますが、データが不十分な場合は、現状の設備に対して改めてIQ/OQ/PQを実施し、適格性を確認する必要があります。これを機に現状の図面化や校正を行うことをお勧めします。
- Q3. バリデーション業務はすべて外部委託してもよいですか?
- 実務作業を外部の専門業者に委託することは可能です。しかし、バリデーション計画の承認や結果の判定、最終的な品質責任は、製造所の製造業者(貴社)が負う必要があります。丸投げにするのではなく、内容を理解し管理することが重要です。
- Q4. OQとPQの違いがよく分かりません。
- OQは「機器単体の機能確認(無負荷)」、PQは「製造プロセス全体としての性能確認(実負荷)」と捉えると分かりやすいでしょう。例えば、インキュベーター単体で温度が出るか確認するのがOQ、実際に培養容器を入れて温度分布を見るのがPQです。
- Q5. バリデーション実施中に不合格(逸脱)が出たら、計画からやり直しですか?
- 必ずしも最初からやり直しではありません。逸脱の原因を特定し、修正(修理や調整)を行った上で、影響を受けた範囲の試験を再実施(再テスト)します。その経緯を逸脱報告書として記録に残すことが適正な手順です。
<script type="application/ld+json">
{
"@context": "https://schema.org",
"@type": "FAQPage",
"mainEntity": [
{
"@type": "Question",
"name": "施設バリデーションにはどのくらいの期間が必要ですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "施設の規模や複雑さによりますが、新規CPCの場合、計画策定からPQ完了まで数ヶ月から半年程度かかることが一般的です。DQは設計段階で行い、IQ/OQは施工完了後、PQは運用開始前に行うため、建設スケジュールと密接に連携する必要があります。"
}
},
{
"@type": "Question",
"name": "既存の施設でバリデーション記録がない場合、どうすればよいですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "「回顧的バリデーション(レトロスペクティブ・バリデーション)」という手法がありますが、データが不十分な場合は、現状の設備に対して改めてIQ/OQ/PQを実施し、適格性を確認する必要があります。これを機に現状の図面化や校正を行うことをお勧めします。"
}
},
{
"@type": "Question",
"name": "バリデーション業務はすべて外部委託してもよいですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "実務作業を外部の専門業者に委託することは可能です。しかし、バリデーション計画の承認や結果の判定、最終的な品質責任は、製造所の製造業者(貴社)が負う必要があります。丸投げにするのではなく、内容を理解し管理することが重要です。"
}
},
{
"@type": "Question",
"name": "OQとPQの違いがよく分かりません。",
"acceptedAnswer": {
"@type": "Answer",
"text": "OQは「機器単体の機能確認(無負荷)」、PQは「製造プロセス全体としての性能確認(実負荷)」と捉えると分かりやすいでしょう。例えば、インキュベーター単体で温度が出るか確認するのがOQ、実際に培養容器を入れて温度分布を見るのがPQです。"
}
},
{
"@type": "Question",
"name": "バリデーション実施中に不合格(逸脱)が出たら、計画からやり直しですか?",
"acceptedAnswer": {
"@type": "Answer",
"text": "必ずしも最初からやり直しではありません。逸脱の原因を特定し、修正(修理や調整)を行った上で、影響を受けた範囲の試験を再実施(再テスト)します。その経緯を逸脱報告書として記録に残すことが適正な手順です。"
}
}
]
}
</script>